For some strange reason, the conventional FASTA contains a sequence that breaks at every 70-100 letters and creates an indentation. This is a major problem if we want to use computational tools to help us analyze genetic information.
To avoid spending too much time re-formatting every FASTA sequence from PubMed, I wrote a Python script to do so. For this example, I used the period gene of Drosophila melanogaster from Flybase. It was my gene of interest during my PhD research.
If you want a step-by-step tutorial on how to write this script, click here.
You will need to download/clone this folder, and in command line, route to this folder using the cd command.
git clone https://github.com/ying-li-python/fasta-fix.git
cd fasta-fix
Example FASTA:
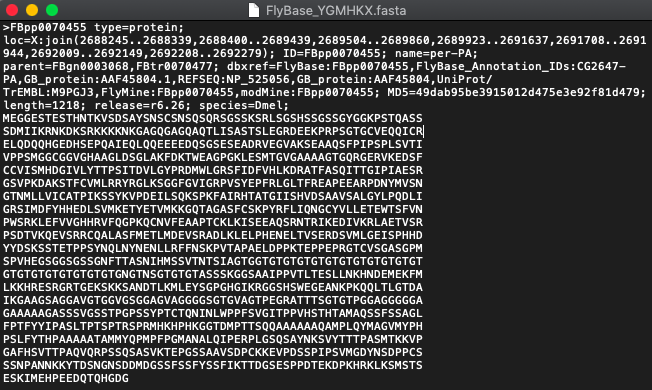
Add a FASTA file in the fasta-fix folder for you to fix. In this case, the file is FlyBase_YGMHKX.fasta.
Using a text or code editor, open fasta_fix.py. I highly recommend Visual Studio.
Replace the FASTA file path to your own.
fastafile = open("FlyBase_YGMHKX.fasta", 'r')
Now that you finished setting up, you are ready to run the the script in command line. Make sure your directory is still in fasta-fix folder.
python fasta_fix.py
Your script will generate a new FASTA file named output.fasta in the same folder. And you're done!
Output:
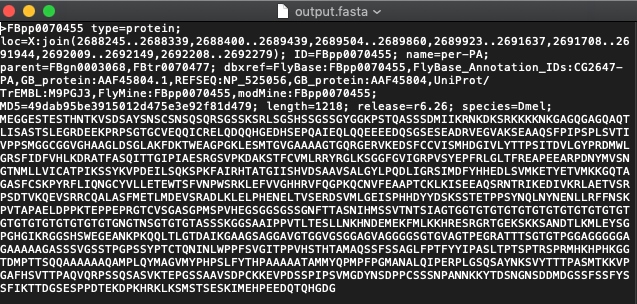
For this script, we created a for-loop, set conditions (if else statements), and used .split() and .close() function.
Ying Li